Epistemic status: This is not medical advice. Pharmacological speculations of a high-schooler, informed by studies done mainly on rodents. Pls don't kill yourself by doing anything suggested in this post, a lot of these substances and combinations of them can be severely dangerous.
TL;DR
An MDMA analogue like 5-MAPB or 4-MMC with low-dose selegiline seems to be the combination with the best MDMA-likeness to neurotoxicity ratio. (targets the two main mechanisms (if hyperthermia is physically avoided) - toxic metabolites and serotonergic neurotoxicity caused by simultaneous dopamine release). Taking selegiline should have the additional effect of a longer/stronger dopaminergic effect due to slowed dopamine degradation. Vitamin C, vitamin E, ALA, and agmatine likely provide further neuroprotection. Antipsychotics like clozapine are an effective measure for MDMA-overdose-caused hyperthermia (only as an emergency measure; otherwise it would dull the effects significantly).
why selegiline
Selegiline is a drug prescribed for Parkinson's disease and depression (for which the patch form - EMSAM - is used in the US).
Mechanism In a 1995 study, it was shown using selegiline and the SSRI fluoxetine, that damage to neuronal membranes from MDMA stems largely from the uptake of dopamine by serotonin transporters, thus causing dopamine to accumulate in serotonergic neurons, where it is broken down mainly by MAO-B (whereas elsewhere, it's metabolised mostly by MAO-A). This deamination by MAO-B creates hydrogen peroxide, which is claimed to be responsible for much of MDMA's neurotoxicity.
Selegiline pharmacology Selegiline is an MAOi; it inhibits the enzymes that metabolise monoamines, such as dopamine, serotonin, norepinephrine, and trace amines, increasing their intersynaptic concentration. Selegiline, along with safinamide and rasagiline, is selective for the MAO-B subtype at certain dosages. The MAO-B enzyme metabolises mostly beta-phenethylamine and dopamine, though it has been found, that MAO-A is mostly responsible for dopamine breakdown, however, it seems likely that it is indeed MAO-B which metabolises dopamine in serotonergic nerve terminals. In addition, there seems to be evidence that MAO-B metabolises serotonin in serotonergic neurons as well, also producing hydrogen peroxide. Selegiline, as well as the beta-phenethylamine it increases (the "endogenous amphetamine"), are agonists of TAAR1. This is also a target of amphetamine, probably being responsible for part of its action. TAAR1 agonism may be responsible for selegiline's catecholaminergic activity enhancer (CAE) activity.[1] In addition, selegiline (especially orally) is metabolised into the levorotary forms of amphetamine and methamphetamine, which act as norepinephrine and dopamine releasing agents, though much weaker in terms of dopamine release than their dextrorotary "conventional" counterparts (detroamphetamine and dextromethamphetamine).[2] Selegiline's CAE effect, amphetamine metabolites, and decreased dopamine metabolism suggest it might enhance MDMA's effect, even in the absence of functional MAO-A inhibition, though it seems from the above-linked studies, that for rodents no significant additional hyperthermia or head-twitch response is observed. This might be due to the fact that most dopamine is metabolised by MAO-A in blood platelets, rather than MAO-B in serotonergic neurons, and thus only the small fraction of dopamine breakdown, that is responsible for neurotoxic effects, is targeted by selegiline administration. Still, it is a potentially risky practice to add selegiline to MDMA use, with possible individual variation in drug metabolism causing MAO-A inhibition at dosages that usually only inhibit MAO-B. Therefore, low dosages of <10 mg, maybe 5 mg, are more reasonable.
Evidence Aside from the Sprague 1995 study linked above, which showed that selegiline (also called Deprenyl) alleviated markers of neurotoxicity in MDMA-exposed rats, in vitro culture had significantly reduced free radical formation when administered selegiline before MDMA, and MAO-B deficient rats where shown to have no serotonin depletion from MDMA (indicating lacking damage to serotonergic neurons - the main targets of MDMA neurotoxicity). Another study from 2007 showed MAO-B dependent damage to mitochondria (including their DNA), alleviated by selegiline. Notably, I didn't find any study testing the combination in humans, nor any anecdotal experience report.
Selectivity/dosage: At dosages of <10 mg/day (orally), selegiline is selective for MAO-B, thus not posing a significant risk of serotonin syndrome[3] (BUT maybe your body works differently and is highly sensitive). If you want to try this potentially dangerous combination, ideally microdose the selegiline at dosages below 5 mg. Patches (EMSAM) might theoretically be useful due to a lower amount of metabolites (reported 70% reduction), but they are risky due to their much higher bioavailability, up to 50-fold, and thus simultaneous MAO-A inhibition at normal dosages (which is the goal of the patches, intended as antidepressants).[4] In addition, it releases the drug slowly over the course of a day, making it less predictable when planning a specific dosage in combination with MDMA.
safety of selected MDMA analogues
Which pharmacological aspects of MDMA do we look for? Besides the dopamine and norepinephrine release and reuptake inhibition, which causes stimulation and euphoria, the specifics of MDMA's effects are likely due to interaction with serotonin receptors. However, it is not entirely clear how serotonin release leads to prosocial, enactogenic effects. Experiments with receptor antagonists have shown that the psychedelic receptor 5-HT2A (which modulates dopamine and prolactin release) is relevant for this effect, as well as 5-HT1B and 5-HT1A, which has downstream effects on increased oxytocin release.[5] Notably, it not only indirectly stimulates but also directly binds to, as a partial agonist, to these receptors, in addition to 5-HT2B and the sigma receptors, which might be responsible for some of its effects. It also binds to alpha-adrenergic receptors, possibly contributing to its anxiolytic effects.[6] Hormonally, MDMA has been shown to elevate the neuropeptides oxytocin and vasopressin, as well as the steroids DHEA and cortisol. The increase in oxytocin seems to be correlated with prosocial effects, and the increase in DHEA with euphoric ones[6] (maybe one should try to supplement DHEA or intranasal oxytocin?). The hormonal effects are, however, much less studied in MDMA analogues than in MDMA itself, so I'll focus on the activity as a triple monoamine releaser and serotonergic receptor activity instead.
Analogues The four following MDMA analogues belong into the classes of benzofurans (5-MAPB and 6-APB) and cathinones (4-MMC and 3-MMC), meaning their cores are different from that of MDMA, and thus form different metabolites. All of them are triple monoamine releasing agents and (except for 5-MAPB) their reuptake inhibitor - leading to increased extracellular concentration of serotonin, dopamine and norepinephrine (noradrenaline). At the same time, all have affinity to the above-mentioned serotonin receptors, mostly as partial agonists (just as MDMA). Importantly, they have none of the metabolites of MDMA, such as alpha-methyldopamine, HHA or HHMA, which are neurotoxic through their auto-oxidation into quinones and their binding to gluthanione, forming neurotoxic thioether conjugates.[7] Therefore, unless there are yet-to-be-discovered neurotoxic metabolites of these drugs, their damage must come primarily from hyperthermia, dopamine breakdown by MAO-B, or RNS formation, which can be alleviated with the methods described in the section below.
5-MAPB It is metabolised into 5-APB (which is a very similar MDMA analogue, however with stronger 5-HT2A agonist and thus likely stronger psychedelic effects), which is subsequently metabolised into 3-carboxymethyl-4-hydroxy amphetamine, and another product is 3-carboxymethyl-4-hydroxy methamphetamine.[8] No catechols or MDA are formed, and so the conversion to quinones or thioether conjugates doesn't happen. However, 5-APB is a potent agonist of the 5-HT2B receptor[9], which might lead to cardiotoxicity with regular use[10] (not recommened for neurotoxicity and monoamine depletion reasons anyway). It has a halflife of 6.5 hours.[11] In terms of effects, the potent 5-HT1B agonism[12] might make this a particularly empathogenic/pro-social compound.[13] However, the lack of direct serotonin 1A receptor activity might make it less oxytocin-releasing, though this mighe be alleviated by stronger serotonin releasing activity (but lower NE activity), and thus indirect 5-HT1A agonism[14], as well as the metabolite 5-APB's activity at the receptor. The lower norepinephrine-releasing activity makes this compound likely less stimulating. 5-MAPB has been found to create hyperthermia (and hypertension, tremor and convulsions) in humans[15], though this is likely to be a case of an overdose. In rat liver cells, 5-MAPB and it's metabolite 5-APB have been shown to cause cell death (cytotoxicity), greater than MDMA.[16]
The Borax combo, as well as 5-MAPB and MDAI, have been advertised as non-neurotoxic alternatives to MDMA.[1][2][5] However, 5-MAPB has subsequently been found to be a serotonergic neurotoxin in rodents similarly to MDMA.[5] It is thought that the serotonergic neurotoxicity of MDMA and related drugs may be dependent on simultaneous induction of serotonin and dopamine release, as combination of a non-neurotoxic serotonin releasing agent like MDAI or MMAI with amphetamine results in serotonergic neurotoxicity similar to that of MDMA.[8][21][22][23] Besides the case of simultaneous induction of serotonin and dopamine release, serotonergic psychedelics (i.e., serotonin 5-HT2 receptor agonists) have been found to augment MDMA-induced striatal dopamine release and serotonergic neurotoxicity in rodents as well.
Confirming the hypothesis of neurotoxicity largely stemming from dopamine breakdown in serotonergic nerves following simultaneous serotonin and dopamine release, such that low-dose selegiline would be protective. If this is the main mechanism of 5-MAPB neurotoxicity, as this suggests, MDMA shouldn't be much worse in comparison, and both should be basically equivalent in terms of their toxicity in combination with an MAO-B inhibitor. However, it is unclear what the role of neurotoxic MDMA metabolites is - it might contribute to neurotoxicity as well, making analogues such as 5-MAPB safer.
In humans, I found one report of fatal intoxication, though it has been in combination with several other compounds.[18] In rodents, 5-MAPB caused similar serotonin depletion as MDMA.[19]
However, much of the toxicity of 5-MAPB compared to MDMA might be caused by use of the same amount of each compound in these studies, even though 5-MAPB is a cca. 3x stronger monoamine releaser than MDMA at the same dose[20][21]. This might simply mean a way too high dosage has been used, leading to more serotonergic hyperthermia + the SERT-mediated toxic dopamine breakdown by MAO-B. Thus at a 3 times lower dosage, it might be a safer alternative to MDMA, especially in combination with low-dose selegiline and hypothermic compounds such as agmatine, alleviating most of the remaining neurotoxicity potential.
6-APB It is much stronger than MDMA - 12x more potent than at the dopamine transporter, 6.5x stronger at the noradrenaline transporter, and 2.4x stronger at the serotonin transporter.[22] With this altered ratio of monoamine release, it can be expected to be more akin to a stimulant like methamphetamine in it's effects. In terms of total monoamine increase measured, all tested benzofurans were about 3x more potent than MDA (so about 9x more potent than MDMA[23]), and 6-APB has been shown to be the most potent benzofuran in terms of dopamine release.[24] In addition, 6-APB was found to bind with high affinity to alpha-adrenergic receptors (similar to MDMA - potentially calming), to 5-HT2A receptors (psychedelic), 5-HT1A receptors (oxytocin-mediating), and, most strongly, to 5-HT2B receptors, which poses potential cardiotoxic risks.[25] The alpha-adrenergic and 5-HT2A agonism make 6-APB a potentially more hyperthermia-inducing drug (which can lead to significant damage, but can also relatively simply be avoided by staying in a cool environment). The effects begin within 1-2 hours and last for about 7 hours.[26] It does not form quinone or thioether metabolites, which contribute to MDMA neurotoxicity (the main 6-APB metabolite was 4-carboxymethyl-3-hydroxy amphetamine).[27] No cytotoxic effects have been found in one cell culture study[28]. There exists a report of 6-APB-caused psychosis, though this has been in combination with cannabis.[29] Overall, 6-APB seems like a more stimulant and psychedelic analogue, with little data on neurotoxicity, though by its similarity to 5-APB it can be assumed to have similar cytotoxic oxidative effects, but less so (it has been found to be less toxic to liver cells than 5-APB, the active 5-MAPB metabolite)[30]
4-MMC aka mephedrone Mephedrone a triple monoamine reuptake inhibitor and releasing agent (as is MDMA, though mephedrone is more of a reuptake inhibitor).[31] It is also a strong 5-HT2A agonist, suggesting it might have psychedelic properties (though strangely, it is not a proper hallucinogen). The lack of direct actiity on the 5-HT1A receptor might mean lower oxytocin release. It is a relatively short-acting drug, with effects beginning after 15 minutes, and lasting 2-3 hours (when taken orally). [32] Mephedrone is commonly insufflated, and is reported to have effects similar to MDMA.[31]
It's metabolites are mostly nor-mephedrone, which is psychoactive itself, as a stimulant (DAT and NET inhibition with less SERT inhibition), DHMMC (which has a similar but weaker profile), and several mostly inactive metabolites like 5-hydroxy-mephedrone.[33] Again, no quinones or thioethers are produced, and none of the studied metabolites has been shown to have neurotoxic properties. Interestingly the article "Clinical Pharmacology of the Synthetic Cathinone Mephedrone"[34] from 2017 reports:
Regarding the possible long-term toxicity of mephedrone, the fact that the drug possesses structural and pharmacological similarities to MDMA, amphetamines, and cathinone suggests the likelihood that repeated and/or prolonged use produces similar consequences on neurochemical and neuropsychological function. From the limited results to date, it should be pointed out that repeated mephedrone administration in experimental animals has not shown evidence of neurotoxicity to monoaminergic systems in the brain [42, 88–91[35]].
One study reports decreased serotonin transporter function in rats administered 4-MMC, but the rats were purposefully kept in a high-temperature environment.[36] Mephedrone induces hyperthermia[37] and potentiates the neurotoxicity of methamphetamine and MDMA, but does not itself cause dopaminergic neurotoxicity. This has lead to the conclusion that mephedrone functions atypically at the dopamine transporter[38] (which might possibly be the reason behind its relative non-neurotoxicity). One rat study showed oxidative damage to rat neurons as well as dopamine receptor downregulation.[39] As opposed to MDMA, mephedrone has not been shown to cause microglial activation, thus the pathway leading to RNS damage is likely nonexistent for mephedrone.[40] Cognitive damage (working memory worsening) has been found in mice after "binge-treatment" of mephedrone.[41] There have been some deaths due to mephedrone overdoses.[42]
Overall, mephedrone seems like a surprisingly safer MDMA alternative, if hyperthermia is avoided (many studies showing harm in rodents used elevated ambient temperature). The working memory deficits shown in rats are concerning, but likely a consequence of high dosages and/or hyperthermia.
3-MMC 3-MMC aka metaphedrone is commonly insufflated 3-MMC inhibits the serotonin transporter much less than mephedrone or MDMA, while significantly inhibiting DAT and NET, suggesting a more stimulant, rather than enactogenic effect.[43] However, the 5-HT1 agonism may lead to oxytocin release, leading to empathogenic effects, confirmed by users.[44] It binds strongly to alpha-adrenergic receptors, which might pose vasoconstriction risk, but the lack of 5-HT2A agonism makes the risk of hyperthermia lower.[43] It is capable of producing hyperthermia, it lasts for around an hourwhen insufflated, and is reported to be a weaker version of mephedrone or MDMA in terms of its effects. The main metabolites are 3-methylephedrine and 3-methylnorephedrine, with no known neurotoxic effect.[45] 3-MMC has been shown to create ROS (and RNS), and damage liver cells.[46] Inhibition of the enzyme CYP2D6 has been shown to be protective, suggesting that genetic variations in the expression of this enzyme may affect the toxicity of 3-MMC use, with "extensive and ultrarapid metabolisers" experiencing significantly more toxicity[45] (which is likely true of MDMA and its other analogues too). Two deaths due to 3-MMC (likely in combination with other drugs) have been reported in Sweden[47], 5 severe poisonings in the Netherlands[48], and adeath following pure 3-MMC exposure in France.[49] Overall, 3-MMC appears to be an inferior alternative to 4-MMC, having a short halflife, shown oxidative stress toxicity, and potentially neurotoxic metabolites.
Taking MDMA has a tradeoff; One gains a euphoric and potentially therapeutic experience, and damages some neurons. The following is a review of the mechanisms by which the cost of MDMA use occurs, and ways to target them.
MDMA induces the release of serotonin, dopamine, acetylcholine and activates histamine receptors, but the main victims of MDMA neurotoxicity appear to be serotonergic (5-HT) axon terminals.[50] There has been a paper claiming dopaminergic neurons are damaged too, but later it was found out that they used meth instead of MDMA.[51] However, some studies do show dopaminergic neurotoxicity of MDMA in rodents as well.[52] Besides damage to axon terminals, damage to the targets of MDMA, the serotonin transporter (SERT) and the dopamine transporter (DAT) has been found, potentially affecting long-term neurotransmitter release and uptake.[53] This post doesn't cover monoamine depletion, which is a short-term effect following MDMA's massive monoamine release, causing the well-known temporary depression and lethargy after MDMA use.
The main mechanisms by which MDMA can be neurotoxic:
Hyperthermia induced by 5−HT2A and β-adrenergic agonism and α-adrenergic receptor-caused vasoconstriction
Creation of ROS from breakdown of dopamine by MAO-B in serotonergic neurons, which damage the neurons' mitochondria[54] (dopamine is broken down into H2O2 (which potentially converts to HO radicals) and catechol metabolites, which auto-oxidise into quinones, both leading to oxidative stress)[55]
Metabolites such as HHMA or α-methyldopamine, auto-oxidising into quinones, creating thioether conjugates with the endogenous antioxidant GSH or NAC [56], that are neurotoxic and cause microglia activation[57] (inflammation, which causes toxicity itself). Or MDA (a metabolite of MDMA):
MDA is metabolized to a-MeDA that can react either with glutathione (GSH) to form 5-(GSH)-a-MeDA or with N-acetylcysteine (NAC) to form 5-(NAC)-a-MeDA, and these compounds might be the main metabolites responsible for the neurotoxic effects of MDMA observed in rats.[58]
4. The microglia-caused inflammation upregulating iNOS, producing NO and subsequently reactive nitrogen species (RNS) including peroxynitrite (which reacts to form nitrotyrosine), which cause oxidative damage to mitochondria[59], cell membranes and proteins (along with the ROS)[60][61]
5. (Rarely, hyponatremia, causing brain swelling. This is more likely with high estrogen exposure)[62]
nonselective MAOIs (and selegiline at high doses) would be very dangerous due to additionally radically increasing intersynaptic serotonin - serotonin syndrome
Analogues that release less dopamine (since the simultaneous release of DA and 5-HT is required for this mechanism of neurotoxicity)
MDAI, MDMAI, MMAI, (R)-MDMA, MBDB, MDAT, 5-APDB, and many more
These, however, likely lack the full subjective effects of MDMA - mostly euphoria
Inhibiting serotonin release (e.g. through SSRIs[76]) or dopamine release (through DRIs[77]) also reduces neurotoxicity, but very likely abolishes or reduces effects
(MDMA reverses the direction of monoamine transporters, so inhibitors of these transporters reduce MDMA effects, even if in the absence of MDMA, they increase intersynaptic concentration of monoamines)
Toxic metabolites:
reducing metabolism
CYP2D6 inhibition (probably dangerous due to higher MDMA levels)
e.g. Bupropion, which also is a DRI (prevents neurotoxicity by previous mechanism)
NOSi, e.g. agmatine[84] (available online) - also reduce hyperthermia
Hyponatremia
Avoid taking estrogen contraceptives or HRT before MDMA
Avoid drinking too much plain water
Antiestrogens
Vitamin E acts as an antiestrogen[85] and antioxidant. It has proven benefits for MDMA hepatotoxicity[86], is depleted by MDMA use, and more neurotoxic damage occurs with its deficiency[87]
ALC administration was found to reduce MDMA-induced protein carbonyl formation (a marker of oxidative protein damage), decrease the incidence of mitochondrial DNA (mtDNA) deletions, and improve the expression of key mitochondrial respiratory chain components (such as subunits of Complex I and Complex IV) [93]
Potentially neuroprotective drugs
modafinil - a DAT inhibitor, shown protective in combination with nicotine[94]
however, modafinil is a CYP450 inducer and as such would speed up MDMA metabolism, causing faster creation of neurotoxic metabolites
bromantane
it enhances dopamine synthesis through tyrosine hydroxylase and DOPA decarboxylase upragulation[95], so it might be a useful post-MDMA dopamine replenisher
Cannabis, strangely enough (due to temperature lowering)[67]
studies referenced: 42. Baumann MH, Ayestas Jr MA, Partilla JS, et al. (2012) The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37:1192–1203 88. Angoa-Pe´rez M, Kane MJ, Francescutti DM, et al. (2012) Mephedrone, an abused psychoactive component of ‘bath salts’ and methamphetamine congener, does not cause neurotoxicity to dopamine nerve endings of the striatum. J Neurochem 120:1097–1107 89. 89. Angoa-Pe´rez M, Kane MJ, Briggs DI, et al. (2013) Mephedrone does not damage dopamine nerve endings of the striatum, but enhances the neurotoxicity of methamphetamine, amphetamine, and MDMA. J Neurochem 125:102–110 90. 90. den Hollander B, Rozov S, Linden AM, et al. (2013) Long-term cognitive and neurochemical effects of “bath salt” designer drugs methylone and mephedrone. Pharmacol Biochem Behav 103:501–509 91. 91. Shortall SE, Green AR, Fone KC, et al. (2016) Caffeine alters the behavioural and body temperature responses to mephedrone without causing long-term neurotoxicity in rats. J Psychopharmacol 30:698–706
The simultaneous large release of dopamine and serotonin causes the serotonin transporter (SERT) to take up some of the dopamine and transport it into the serotonergic nerve, where it is broken down by MAO-B (usually MAO-A breaks down dopamine and serotonin, but MAO-B is found in the serotonergic neurons and is responsible for the breakdown of the residual monoamines). This creates free radicals that can cause damage to the membranes and mitochondria of neurons. That's why the inventor of Selegiline, an MAO-B inhibitor, promoted its preventative use as a longevity drug (he did live until the age of 92).
Epistemic status: This is not medical advice. Pharmacological speculations of a high-schooler, informed by studies done mainly on rodents. Pls don't kill yourself by doing anything suggested in this post, a lot of these substances and combinations of them can be severely dangerous.
TL;DR
An MDMA analogue like 5-MAPB or 4-MMC with low-dose selegiline seems to be the combination with the best MDMA-likeness to neurotoxicity ratio. (targets the two main mechanisms (if hyperthermia is physically avoided) - toxic metabolites and serotonergic neurotoxicity caused by simultaneous dopamine release). Taking selegiline should have the additional effect of a longer/stronger dopaminergic effect due to slowed dopamine degradation.
Vitamin C, vitamin E, ALA, and agmatine likely provide further neuroprotection. Antipsychotics like clozapine are an effective measure for MDMA-overdose-caused hyperthermia (only as an emergency measure; otherwise it would dull the effects significantly).
why selegiline
Selegiline is a drug prescribed for Parkinson's disease and depression (for which the patch form - EMSAM - is used in the US).
Mechanism
In a 1995 study, it was shown using selegiline and the SSRI fluoxetine, that damage to neuronal membranes from MDMA stems largely from the uptake of dopamine by serotonin transporters, thus causing dopamine to accumulate in serotonergic neurons, where it is broken down mainly by MAO-B (whereas elsewhere, it's metabolised mostly by MAO-A). This deamination by MAO-B creates hydrogen peroxide, which is claimed to be responsible for much of MDMA's neurotoxicity.
Selegiline pharmacology
Selegiline is an MAOi; it inhibits the enzymes that metabolise monoamines, such as dopamine, serotonin, norepinephrine, and trace amines, increasing their intersynaptic concentration.
Selegiline, along with safinamide and rasagiline, is selective for the MAO-B subtype at certain dosages. The MAO-B enzyme metabolises mostly beta-phenethylamine and dopamine, though it has been found, that MAO-A is mostly responsible for dopamine breakdown, however, it seems likely that it is indeed MAO-B which metabolises dopamine in serotonergic nerve terminals. In addition, there seems to be evidence that MAO-B metabolises serotonin in serotonergic neurons as well, also producing hydrogen peroxide.
Selegiline, as well as the beta-phenethylamine it increases (the "endogenous amphetamine"), are agonists of TAAR1. This is also a target of amphetamine, probably being responsible for part of its action. TAAR1 agonism may be responsible for selegiline's catecholaminergic activity enhancer (CAE) activity.[1]
In addition, selegiline (especially orally) is metabolised into the levorotary forms of amphetamine and methamphetamine, which act as norepinephrine and dopamine releasing agents, though much weaker in terms of dopamine release than their dextrorotary "conventional" counterparts (detroamphetamine and dextromethamphetamine).[2]
Selegiline's CAE effect, amphetamine metabolites, and decreased dopamine metabolism suggest it might enhance MDMA's effect, even in the absence of functional MAO-A inhibition, though it seems from the above-linked studies, that for rodents no significant additional hyperthermia or head-twitch response is observed. This might be due to the fact that most dopamine is metabolised by MAO-A in blood platelets, rather than MAO-B in serotonergic neurons, and thus only the small fraction of dopamine breakdown, that is responsible for neurotoxic effects, is targeted by selegiline administration.
Still, it is a potentially risky practice to add selegiline to MDMA use, with possible individual variation in drug metabolism causing MAO-A inhibition at dosages that usually only inhibit MAO-B. Therefore, low dosages of <10 mg, maybe 5 mg, are more reasonable.
Evidence
Aside from the Sprague 1995 study linked above, which showed that selegiline (also called Deprenyl) alleviated markers of neurotoxicity in MDMA-exposed rats, in vitro culture had significantly reduced free radical formation when administered selegiline before MDMA, and MAO-B deficient rats where shown to have no serotonin depletion from MDMA (indicating lacking damage to serotonergic neurons - the main targets of MDMA neurotoxicity). Another study from 2007 showed MAO-B dependent damage to mitochondria (including their DNA), alleviated by selegiline.
Notably, I didn't find any study testing the combination in humans, nor any anecdotal experience report.
Selectivity/dosage:
At dosages of <10 mg/day (orally), selegiline is selective for MAO-B, thus not posing a significant risk of serotonin syndrome[3] (BUT maybe your body works differently and is highly sensitive). If you want to try this potentially dangerous combination, ideally microdose the selegiline at dosages below 5 mg.
Patches (EMSAM) might theoretically be useful due to a lower amount of metabolites (reported 70% reduction), but they are risky due to their much higher bioavailability, up to 50-fold, and thus simultaneous MAO-A inhibition at normal dosages (which is the goal of the patches, intended as antidepressants).[4] In addition, it releases the drug slowly over the course of a day, making it less predictable when planning a specific dosage in combination with MDMA.
safety of selected MDMA analogues
Which pharmacological aspects of MDMA do we look for?
Besides the dopamine and norepinephrine release and reuptake inhibition, which causes stimulation and euphoria, the specifics of MDMA's effects are likely due to interaction with serotonin receptors. However, it is not entirely clear how serotonin release leads to prosocial, enactogenic effects. Experiments with receptor antagonists have shown that the psychedelic receptor 5-HT2A (which modulates dopamine and prolactin release) is relevant for this effect, as well as 5-HT1B and 5-HT1A, which has downstream effects on increased oxytocin release.[5] Notably, it not only indirectly stimulates but also directly binds to, as a partial agonist, to these receptors, in addition to 5-HT2B and the sigma receptors, which might be responsible for some of its effects. It also binds to alpha-adrenergic receptors, possibly contributing to its anxiolytic effects.[6]
Hormonally, MDMA has been shown to elevate the neuropeptides oxytocin and vasopressin, as well as the steroids DHEA and cortisol. The increase in oxytocin seems to be correlated with prosocial effects, and the increase in DHEA with euphoric ones[6] (maybe one should try to supplement DHEA or intranasal oxytocin?). The hormonal effects are, however, much less studied in MDMA analogues than in MDMA itself, so I'll focus on the activity as a triple monoamine releaser and serotonergic receptor activity instead.
Analogues
The four following MDMA analogues belong into the classes of benzofurans (5-MAPB and 6-APB) and cathinones (4-MMC and 3-MMC), meaning their cores are different from that of MDMA, and thus form different metabolites.
All of them are triple monoamine releasing agents and (except for 5-MAPB) their reuptake inhibitor - leading to increased extracellular concentration of serotonin, dopamine and norepinephrine (noradrenaline). At the same time, all have affinity to the above-mentioned serotonin receptors, mostly as partial agonists (just as MDMA).
Importantly, they have none of the metabolites of MDMA, such as alpha-methyldopamine, HHA or HHMA, which are neurotoxic through their auto-oxidation into quinones and their binding to gluthanione, forming neurotoxic thioether conjugates.[7] Therefore, unless there are yet-to-be-discovered neurotoxic metabolites of these drugs, their damage must come primarily from hyperthermia, dopamine breakdown by MAO-B, or RNS formation, which can be alleviated with the methods described in the section below.
5-MAPB
It is metabolised into 5-APB (which is a very similar MDMA analogue, however with stronger 5-HT2A agonist and thus likely stronger psychedelic effects), which is subsequently metabolised into 3-carboxymethyl-4-hydroxy amphetamine, and another product is 3-carboxymethyl-4-hydroxy methamphetamine.[8] No catechols or MDA are formed, and so the conversion to quinones or thioether conjugates doesn't happen. However, 5-APB is a potent agonist of the 5-HT2B receptor[9], which might lead to cardiotoxicity with regular use[10] (not recommened for neurotoxicity and monoamine depletion reasons anyway).
It has a halflife of 6.5 hours.[11]
In terms of effects, the potent 5-HT1B agonism[12] might make this a particularly empathogenic/pro-social compound.[13] However, the lack of direct serotonin 1A receptor activity might make it less oxytocin-releasing, though this mighe be alleviated by stronger serotonin releasing activity (but lower NE activity), and thus indirect 5-HT1A agonism[14], as well as the metabolite 5-APB's activity at the receptor. The lower norepinephrine-releasing activity makes this compound likely less stimulating.
5-MAPB has been found to create hyperthermia (and hypertension, tremor and convulsions) in humans[15], though this is likely to be a case of an overdose. In rat liver cells, 5-MAPB and it's metabolite 5-APB have been shown to cause cell death (cytotoxicity), greater than MDMA.[16]
From Wikipedia[17]
Confirming the hypothesis of neurotoxicity largely stemming from dopamine breakdown in serotonergic nerves following simultaneous serotonin and dopamine release, such that low-dose selegiline would be protective. If this is the main mechanism of 5-MAPB neurotoxicity, as this suggests, MDMA shouldn't be much worse in comparison, and both should be basically equivalent in terms of their toxicity in combination with an MAO-B inhibitor. However, it is unclear what the role of neurotoxic MDMA metabolites is - it might contribute to neurotoxicity as well, making analogues such as 5-MAPB safer.
In humans, I found one report of fatal intoxication, though it has been in combination with several other compounds.[18]
In rodents, 5-MAPB caused similar serotonin depletion as MDMA.[19]
However, much of the toxicity of 5-MAPB compared to MDMA might be caused by use of the same amount of each compound in these studies, even though 5-MAPB is a cca. 3x stronger monoamine releaser than MDMA at the same dose[20][21]. This might simply mean a way too high dosage has been used, leading to more serotonergic hyperthermia + the SERT-mediated toxic dopamine breakdown by MAO-B. Thus at a 3 times lower dosage, it might be a safer alternative to MDMA, especially in combination with low-dose selegiline and hypothermic compounds such as agmatine, alleviating most of the remaining neurotoxicity potential.
6-APB
It is much stronger than MDMA - 12x more potent than at the dopamine transporter, 6.5x stronger at the noradrenaline transporter, and 2.4x stronger at the serotonin transporter.[22] With this altered ratio of monoamine release, it can be expected to be more akin to a stimulant like methamphetamine in it's effects. In terms of total monoamine increase measured, all tested benzofurans were about 3x more potent than MDA (so about 9x more potent than MDMA[23]), and 6-APB has been shown to be the most potent benzofuran in terms of dopamine release.[24]
In addition, 6-APB was found to bind with high affinity to alpha-adrenergic receptors (similar to MDMA - potentially calming), to 5-HT2A receptors (psychedelic), 5-HT1A receptors (oxytocin-mediating), and, most strongly, to 5-HT2B receptors, which poses potential cardiotoxic risks.[25] The alpha-adrenergic and 5-HT2A agonism make 6-APB a potentially more hyperthermia-inducing drug (which can lead to significant damage, but can also relatively simply be avoided by staying in a cool environment).
The effects begin within 1-2 hours and last for about 7 hours.[26]
It does not form quinone or thioether metabolites, which contribute to MDMA neurotoxicity (the main 6-APB metabolite was 4-carboxymethyl-3-hydroxy amphetamine).[27]
No cytotoxic effects have been found in one cell culture study[28]. There exists a report of 6-APB-caused psychosis, though this has been in combination with cannabis.[29]
Overall, 6-APB seems like a more stimulant and psychedelic analogue, with little data on neurotoxicity, though by its similarity to 5-APB it can be assumed to have similar cytotoxic oxidative effects, but less so (it has been found to be less toxic to liver cells than 5-APB, the active 5-MAPB metabolite)[30]
4-MMC aka mephedrone
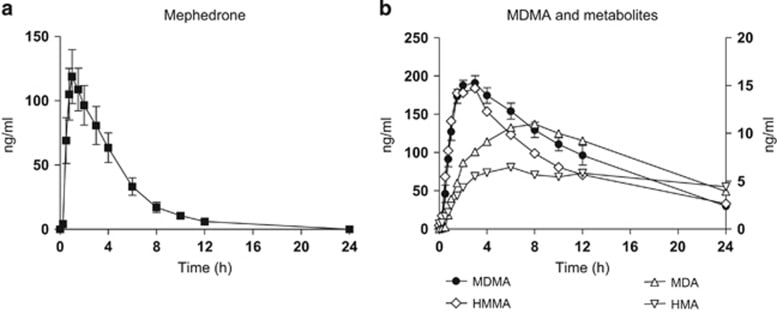 [32]
[32]
Mephedrone a triple monoamine reuptake inhibitor and releasing agent (as is MDMA, though mephedrone is more of a reuptake inhibitor).[31] It is also a strong 5-HT2A agonist, suggesting it might have psychedelic properties (though strangely, it is not a proper hallucinogen). The lack of direct actiity on the 5-HT1A receptor might mean lower oxytocin release.
It is a relatively short-acting drug, with effects beginning after 15 minutes, and lasting 2-3 hours (when taken orally).
Mephedrone is commonly insufflated, and is reported to have effects similar to MDMA.[31]
It's metabolites are mostly nor-mephedrone, which is psychoactive itself, as a stimulant (DAT and NET inhibition with less SERT inhibition), DHMMC (which has a similar but weaker profile), and several mostly inactive metabolites like 5-hydroxy-mephedrone.[33] Again, no quinones or thioethers are produced, and none of the studied metabolites has been shown to have neurotoxic properties.
Interestingly the article "Clinical Pharmacology of the Synthetic Cathinone Mephedrone"[34] from 2017 reports:
One study reports decreased serotonin transporter function in rats administered 4-MMC, but the rats were purposefully kept in a high-temperature environment.[36]
Mephedrone induces hyperthermia[37] and potentiates the neurotoxicity of methamphetamine and MDMA, but does not itself cause dopaminergic neurotoxicity. This has lead to the conclusion that mephedrone functions atypically at the dopamine transporter[38] (which might possibly be the reason behind its relative non-neurotoxicity).
One rat study showed oxidative damage to rat neurons as well as dopamine receptor downregulation.[39]
As opposed to MDMA, mephedrone has not been shown to cause microglial activation, thus the pathway leading to RNS damage is likely nonexistent for mephedrone.[40] Cognitive damage (working memory worsening) has been found in mice after "binge-treatment" of mephedrone.[41] There have been some deaths due to mephedrone overdoses.[42]
Overall, mephedrone seems like a surprisingly safer MDMA alternative, if hyperthermia is avoided (many studies showing harm in rodents used elevated ambient temperature). The working memory deficits shown in rats are concerning, but likely a consequence of high dosages and/or hyperthermia.
3-MMC
3-MMC aka metaphedrone is commonly insufflated
3-MMC inhibits the serotonin transporter much less than mephedrone or MDMA, while significantly inhibiting DAT and NET, suggesting a more stimulant, rather than enactogenic effect.[43] However, the 5-HT1 agonism may lead to oxytocin release, leading to empathogenic effects, confirmed by users.[44] It binds strongly to alpha-adrenergic receptors, which might pose vasoconstriction risk, but the lack of 5-HT2A agonism makes the risk of hyperthermia lower.[43] It is capable of producing hyperthermia, it lasts for around an hourwhen insufflated, and is reported to be a weaker version of mephedrone or MDMA in terms of its effects. The main metabolites are 3-methylephedrine and 3-methylnorephedrine, with no known neurotoxic effect.[45] 3-MMC has been shown to create ROS (and RNS), and damage liver cells.[46] Inhibition of the enzyme CYP2D6 has been shown to be protective, suggesting that genetic variations in the expression of this enzyme may affect the toxicity of 3-MMC use, with "extensive and ultrarapid metabolisers" experiencing significantly more toxicity[45] (which is likely true of MDMA and its other analogues too).
Two deaths due to 3-MMC (likely in combination with other drugs) have been reported in Sweden[47], 5 severe poisonings in the Netherlands[48], and adeath following pure 3-MMC exposure in France.[49]
Overall, 3-MMC appears to be an inferior alternative to 4-MMC, having a short halflife, shown oxidative stress toxicity, and potentially neurotoxic metabolites.
Taking MDMA has a tradeoff; One gains a euphoric and potentially therapeutic experience, and damages some neurons. The following is a review of the mechanisms by which the cost of MDMA use occurs, and ways to target them.
MDMA induces the release of serotonin, dopamine, acetylcholine and activates histamine receptors, but the main victims of MDMA neurotoxicity appear to be serotonergic (5-HT) axon terminals.[50] There has been a paper claiming dopaminergic neurons are damaged too, but later it was found out that they used meth instead of MDMA.[51] However, some studies do show dopaminergic neurotoxicity of MDMA in rodents as well.[52] Besides damage to axon terminals, damage to the targets of MDMA, the serotonin transporter (SERT) and the dopamine transporter (DAT) has been found, potentially affecting long-term neurotransmitter release and uptake.[53]
This post doesn't cover monoamine depletion, which is a short-term effect following MDMA's massive monoamine release, causing the well-known temporary depression and lethargy after MDMA use.
The main mechanisms by which MDMA can be neurotoxic:
Or MDA (a metabolite of MDMA):
4. The microglia-caused inflammation upregulating iNOS, producing NO and subsequently reactive nitrogen species (RNS) including peroxynitrite (which reacts to form nitrotyrosine), which cause oxidative damage to mitochondria[59], cell membranes and proteins (along with the ROS)[60][61]
5. (Rarely, hyponatremia, causing brain swelling. This is more likely with high estrogen exposure)[62]
What can be done:
CYP2D6 inhibition(probably dangerous due to higher MDMA levels)https://pmc.ncbi.nlm.nih.gov/articles/PMC10487936/
https://en.wikipedia.org/wiki/Levomethamphetamine#Pharmacology
https://www.benchchem.com/pdf/Technical_Support_Center_Optimizing_Selegiline_Dosage_for_Selective_MAO_B_Inhibition_In_Vivo.pdf
https://pmc.ncbi.nlm.nih.gov/articles/PMC2656289/#sec4
https://pmc.ncbi.nlm.nih.gov/articles/PMC4678620/
https://shaunlacob.com/wp-content/uploads/2020/12/DC-MDMA.pdf
https://pmc.ncbi.nlm.nih.gov/articles/PMC2698942/
https://pubmed.ncbi.nlm.nih.gov/25471293/
https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.13128
https://en.wikipedia.org/wiki/5-HT2B_receptor#Clinical_significance
https://www.sciencedirect.com/science/article/abs/pii/S0196064416300038
https://patents.google.com/patent/US20230150963A1/en
https://pmc.ncbi.nlm.nih.gov/articles/PMC8990025/#S2
https://openaccess.sgul.ac.uk/id/eprint/108925/1/Combined_in_vitro_and_in_silico_approaches.pdf
https://pubmed.ncbi.nlm.nih.gov/27156124/
https://pubmed.ncbi.nlm.nih.gov/27291301/ or https://www.wellesu.com/10.1002/jat.3351
https://en.wikipedia.org/wiki/Borax_combo#Neurotoxicity
https://pubmed.ncbi.nlm.nih.gov/38649548/
https://www.abstractsonline.com/pp8/#!/10619/presentation/67382
https://www.researchgate.net/figure/Dose-response-effects-of-MDMA-5-MAPB-and-6-MAPB-to-induce-release-of-HMPP-via-DAT_fig3_344040290
https://pubmed.ncbi.nlm.nih.gov/38272669/
https://pmc.ncbi.nlm.nih.gov/articles/PMC7686291/table/T1/
https://en.wikipedia.org/wiki/3,4-Methylenedioxyamphetamine#Pharmacology
https://pmc.ncbi.nlm.nih.gov/articles/PMC7686291/#S15
https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.13128
https://wellesu.com/10.1016/j.drugalcdep.2015.10.011
https://www.researchgate.net/publication/272843882_Metabolic_fate_mass_spectral_fragmentation_detectability_and_differentiation_in_urine_of_the_benzofuran_designer_drugs_6-APB_and_6-MAPB_in_comparison_to_their_5-isomers_using_GC-MS_and_LC-HR-MSn_techn
https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.13128
https://pmc.ncbi.nlm.nih.gov/articles/PMC3770991/
https://www.springermedizin.de/emerging-club-drugs-5-2-aminopropyl-benzofuran-5-apb-is-more-tox/50551032
https://wellesu.com/10.1007/7854_2016_61
https://pmc.ncbi.nlm.nih.gov/articles/PMC5026738
https://www.mdpi.com/1422-0067/26/15/7656#Phase_I_Metabolites
https://wellesu.com/10.1007/7854_2016_61
studies referenced:
42. Baumann MH, Ayestas Jr MA, Partilla JS, et al. (2012) The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37:1192–1203
88. Angoa-Pe´rez M, Kane MJ, Francescutti DM, et al. (2012) Mephedrone, an abused psychoactive component of ‘bath salts’ and methamphetamine congener, does not cause neurotoxicity to dopamine nerve endings of the striatum. J Neurochem 120:1097–1107 89.
89. Angoa-Pe´rez M, Kane MJ, Briggs DI, et al. (2013) Mephedrone does not damage dopamine nerve endings of the striatum, but enhances the neurotoxicity of methamphetamine, amphetamine, and MDMA. J Neurochem 125:102–110 90.
90. den Hollander B, Rozov S, Linden AM, et al. (2013) Long-term cognitive and neurochemical effects of “bath salt” designer drugs methylone and mephedrone. Pharmacol Biochem Behav 103:501–509 91.
91. Shortall SE, Green AR, Fone KC, et al. (2016) Caffeine alters the behavioural and body temperature responses to mephedrone without causing long-term neurotoxicity in rats. J Psychopharmacol 30:698–706
https://pmc.ncbi.nlm.nih.gov/articles/PMC3200001/#sec14
https://pmc.ncbi.nlm.nih.gov/articles/PMC5771050/#r42
https://pmc.ncbi.nlm.nih.gov/articles/PMC3604033/
https://pubmed.ncbi.nlm.nih.gov/25817894/
https://pmc.ncbi.nlm.nih.gov/articles/PMC5771050/#r42
https://pubmed.ncbi.nlm.nih.gov/23099177/
https://www.erowid.org/chemicals/4_methylmethcathinone/4_methylmethcathinone_health1.shtml
https://wellesu.com/10.1016/j.neuropharm.2017.07.026
https://erowid.org/experiences/subs/exp_3Methylmethcathinone.shtml
https://pmc.ncbi.nlm.nih.gov/articles/PMC10972361/#sec3-medicina-60-00466
https://pubmed.ncbi.nlm.nih.gov/31468101/
https://pubmed.ncbi.nlm.nih.gov/25422862/
https://pubmed.ncbi.nlm.nih.gov/35752518/
https://www.sciencedirect.com/science/article/abs/pii/S2352007816302359
https://pmc.ncbi.nlm.nih.gov/articles/PMC2505334/
https://en.wikipedia.org/wiki/Retracted_article_on_dopaminergic_neurotoxicity_of_MDMA
https://www.springermedizin.de/progression-and-persistence-of-neurotoxicity-induced-by-mdma-in-/25643874
https://pmc.ncbi.nlm.nih.gov/articles/PMC3955110
https://pmc.ncbi.nlm.nih.gov/articles/PMC6672671/
https://pmc.ncbi.nlm.nih.gov/articles/PMC2911494/
https://pmc.ncbi.nlm.nih.gov/articles/PMC2698942/
https://pmc.ncbi.nlm.nih.gov/articles/PMC3930364/#s11
https://pubmed.ncbi.nlm.nih.gov/25096201/
https://pmc.ncbi.nlm.nih.gov/articles/PMC9149009/
https://pmc.ncbi.nlm.nih.gov/articles/PMC2911494/
https://www.mdpi.com/1422-0067/20/5/1242
https://pmc.ncbi.nlm.nih.gov/articles/PMC3769979/
https://pmc.ncbi.nlm.nih.gov/articles/PMC3492978/
https://pubmed.ncbi.nlm.nih.gov/8764359/
https://pmc.ncbi.nlm.nih.gov/articles/PMC6740548/
https://www.nature.com/articles/npp2015182
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0009143
https://www.researchgate.net/publication/5867052_Memantine_prevents_MDMA-induced_neurotoxicity
https://www.sciencedirect.com/science/article/abs/pii/030439408990637X
https://pmc.ncbi.nlm.nih.gov/articles/PMC1572911/
https://pmc.ncbi.nlm.nih.gov/articles/PMC399103
https://fse.studenttheses.ub.rug.nl/12564/
https://pubmed.ncbi.nlm.nih.gov/6781712/
The simultaneous large release of dopamine and serotonin causes the serotonin transporter (SERT) to take up some of the dopamine and transport it into the serotonergic nerve, where it is broken down by MAO-B (usually MAO-A breaks down dopamine and serotonin, but MAO-B is found in the serotonergic neurons and is responsible for the breakdown of the residual monoamines). This creates free radicals that can cause damage to the membranes and mitochondria of neurons. That's why the inventor of Selegiline, an MAO-B inhibitor, promoted its preventative use as a longevity drug (he did live until the age of 92).
https://pubmed.ncbi.nlm.nih.gov/7542394/
https://pubmed.ncbi.nlm.nih.gov/19682588/
https://pubmed.ncbi.nlm.nih.gov/10349862/
https://en.wikipedia.org/wiki/CYP2D6#Ligands
https://onlinelibrary.wiley.com/doi/10.1111/jnc.16149
https://en.wikipedia.org/wiki/4-Fluoroamphetamine#Pharmacology
https://pmc.ncbi.nlm.nih.gov/articles/PMC12715756/
https://en.wikipedia.org/wiki/5-MAPB#Pharmacokinetics
reactive nitrogen species
https://pmc.ncbi.nlm.nih.gov/articles/PMC3991030/#sec9
https://pubmed.ncbi.nlm.nih.gov/16091003/
https://pmc.ncbi.nlm.nih.gov/articles/PMC7698444/
https://www.sciencedirect.com/science/article/abs/pii/S0006899302023132
https://pubmed.ncbi.nlm.nih.gov/11170222/
https://pubmed.ncbi.nlm.nih.gov/25096201/
https://pubmed.ncbi.nlm.nih.gov/17467183/
https://pubmed.ncbi.nlm.nih.gov/10619665/
https://fse.studenttheses.ub.rug.nl/12564/1/LST_BC_2015_BMNijhoff.pdf
Acetyl-L-carnitine provides effective in vivo neuroprotection over 3,4-methylenedioximethamphetamine-induced mitochondrial neurotoxicity in the adolescent rat brain - PubMed
https://www.sciencedirect.com/science/article/pii/S0891061821000697
https://www.wellesu.com/10.1007/s10628-005-0057-z