I'm not sure what epigenetics researchers you've been talking to, but my colleagues and I are all interested in the dynamic interplay between epigenetic modalities (DNA methylation, 3D genome architecture, accessibility, histone modifications, transcription factor binding, transcription, proteomics).
The 2020 paper you cite shows what was, for me, a surprisingly gradual rate of decay in CG methylation after knocking out DNMT3a/b. It seems incompatible with "turnover every few days" in most positions, although that could be happening in some locations. We definitely need a much deeper understanding of DNA methylation dynamics and heterogeneity - especially, in my opinion, at individual CG sites at single-cell resolution.
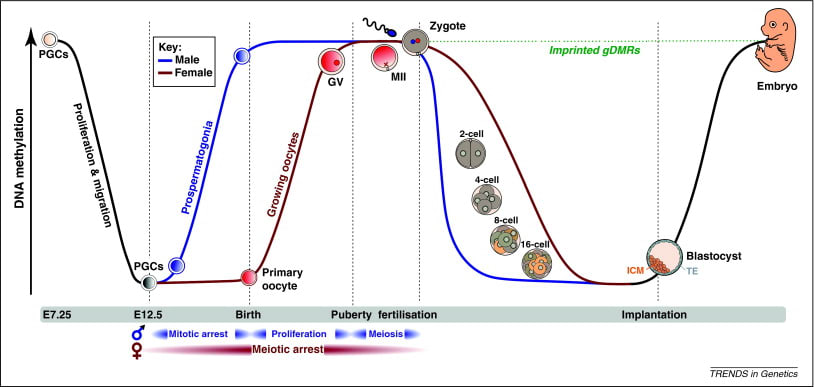
Just for perspective, demethylation can happen much faster, crashing dramatically genome-wide over just a few days during embryonic development.
And the median mRNA half-life has been estimated at 10h[1], compared to the time scales of a week to more than a month measured above. So mRNA seems like a relatively stable layer of the epigenome on the whole.
Another interesting aspect of DNA methylation is that CH methylation (methylation at cytosines outside a CG context) accumulates in long-lived post-mitotic cells, like neurons, myofibers, and placental trophoblast. We know it's functional in neurons, but to my eye, in myofibers and trophoblast, it looks like off-target deposition that correlates with CG deposition, and I wonder if it's just off-target methylation that's not getting cleared. If methylation gets deposited or cleared in an off-target manner, then breakdown in whatever role it locally plays in epigenetic regulation can probably set the rest of the mechanism off-balance, resulting in a gradual slide toward dysregulation over time. My expectation is that aging is the result of an overall "smearing" of epigenetic regulation in which accumulated noise and tail events gradually hamper normal cell function more and more until one system or another suffers a catastrophic failure that cascades through the rest of the body.
It appears that partial reprogramming of stem cells can substantially rejuvenate the epigenetic state. I don't have a link handy but I'll have to write about that sometime. My guess is that it will one day be possible to just reset the epigenetic state of stem cells in non-brain tissues and achieve substantial anti-aging therapies that way. I'm less optimistic about near-term solutions for brain rejuvenation, since neurons are canonically post-mitotic and the evidence for adult neurogenesis seems like it's on shaky ground. But who knows? Maybe we'll figure out a neurorejuvenative therapy that treats Alzheimer's, discover the same treatment works as a prophylactic, and then discover it can be applied generally to improve brain function in middle-aged adults!
At some point, I may write a longer "News and Views" style essay on this topic, and I'll post it on LessWrong if so. I'll also be writing a similar essay on DNA damage and DNA methylation, and I guess I'll post that on here as well if I don't just merge them into the same work.
Probably not the type of reference you were thinking about regarding reprogramming and impact on aging issues but I suspect it's in the area you were thinking. I'm pretty sure it's been mentioned here on LW before in other posts/comments. Interesting idea but implementation is problematic to say the least -- but really hoping someone can figure it out.
Since a lot of this is way beyond my skill sets and knowledge, when you're looking at the dynamic interplay aspect, is that purely internal to the cell or do you also look at the extra-cellular "communications"? If so, are you familiar with the Conboy's plasma dilution experiments?
Our lab focuses on single-cell sequencing based technology development and computational methods. These methods yield a per-cell, sparse snapshot of one or more aspects of cell-intrinsic chromatin or transcriptome state. Some of us work on spatial methods, which allow tagging the profiled cells with a marker of their physical position in a tissue. Some of us also use a variety of computational methods to infer a temporal component from the snapshot data, in a manner analogous to chronophotography.
I don't think our lab's currently working on any inter-cellular communications, but it would be an interesting issue to work on.
Also, I'm just going to put this out there because times are tough in the ol' science funding realm. If anybody wants to donate to our lab, we have the world's best assay for profiling DNA methylation at single-cell resolution, sciMETv3. We can profile hundreds of thousands of individual cells in a single assay, with the cost mainly driven by sequencing. If you're interested in donating to the lab, we could almost certainly arrange that through our university's foundation and explore ideas for projects. We are also actively developing multimodal assays. DM me if interested!
I have a pet lay-speculation that there's a pretty mathematically interesting question here, which hasn't been understood yet. I can't formulate the question clearly, but it's something like: "What sort of thing are these states?" We can abstractly talk about stable states of high-dimensional dynamical systems, but this probably isn't very satisfying or helpful in this context. There's some more practical or concrete or specific things we might want to know about the landscape of possible stable or quasi-stable states for gene regulatory networks, and how they transition, and how one could perturb them.
In the case of epigenetic memory based on freely-diffusing factors, the alternative "stable" states can probably be thought of as long-lived metastable states in "real" stochastic system, which become stable fixed points in the limit as the number of particles N goes to infinity. In models, the switching time often grows exponentially with the number of particles. You may enjoy https://arxiv.org/abs/q-bio/0410003 or https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.115.208101.
For memory based on chemical modifications embedded along the genome, like DNA methylation, there isn't really a "large N" limit to take, and in my view things are less settled. You may enjoy https://pubmed.ncbi.nlm.nih.gov/17512413/ or (shameless plug) https://www.science.org/doi/10.1126/science.adg3053
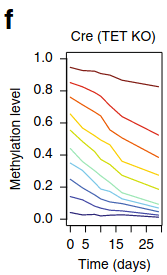
The mistake: methyl groups usually do not stick around long-term; they turn over regularly. Here are two studies which measured the turnover. Turnover timescale varies by location on the DNA strand, but turnover every few days is typical.
I don't know if someone else was making this particular mistake. I certainly find it quite tricky to describe these things with language unless I am extremely careful. I am still confused, but I do find your suggested pathway via transposons more plausible, and the story for why transposons fits into my head. I don't think a correct story for why not epigenetic "bit-flips" currently fits properly into my head, and that correct story would have to be created in order to convince other people. Ideally, some testable predictions[1].
But if the methyl groups are instead part of a dynamic equilibrium, especially with high redundancy (and therefore stability), then that's a whole different situation.
I don't understand why this has to be such a dichotomy? What I hear you say is that these dynamic equilibria are too stable to be a root cause of aging. I think this is true in the case of X-inactivation[2], which is redundant across most of the X-chromosome. But as far as I can tell, the number of redundant parts that make up one "unit" that can bit-flip lies on a spectrum. Example: one methylated C is probably too unstable to encode something. But what if I have a local cluster of five of them where each one reinforces each other (because enzymes that change the methylation often check the methylation status of close-by C's)? That's the point of CpG islands, right? So some of these CpG islands are probably more stable than others, and some could be just stable/unstable enough to be a root cause of aging, no?
For example, my guess would be that the transposon story and the accumulating epigenetic chaos story make different predictions about which methylations would serve as an aging clock and how well. They would also predict different things if we compare aging in different species, because transposons and mitochondria are (quite?) universal in eukaryotes, while epigenetics between eukaryotes is quite different. ↩︎
My understanding is X-inactivation almost never bit-flips in adults. If it were unstable on the timescale of years, you'd sometimes see one white hair in a spot where a cat has black hair, for example. Maybe it is more common in cells with faster turnover? ↩︎
I don't understand why this has to be such a dichotomy?
Good question. I intentionally didn't get into the details in the post, but happy to walk through it in the comments.
Imagine some not-intended-to-be-realistic simplified toy organism with two cell types, A and B. This simplified toy organism has 1001 methyl modification sites which control the cell's type; each site pushes the cell toward either type A or type B (depending on whether the methyl is present or not), and whatever state the majority push toward, that's the type the cell will express.
Under that setup, what do our two mental models look like?
The no-methyl-turnover model would say: well, somewhere upstream in development the methyl groups almost-all lock in to the same cell state, let's say type A. And then the cell (and its descendants, barring some programmed change) stays in that state indefinitely, because the methyl groups stay in that state indefinitely. Maybe aging involves the methyl groups slowly flipping as entropy does its work, until eventually the majority-A is lost, and the cell switches to the "wrong" type.
The dynamic steady state model would instead say: even if a few of the 1001 methyl groups flip to the "wrong" state at some point, they'll just be flipped back soon after. Something is pushing the methyls back toward the consensus value. That means there's no mechanism for the memory to slowly "decay"; a state change would only happen if some big shock makes a whole bunch of the methyl groups flip at the same time. So under the dynamic model, there's no gradual loss of memory; there's just a(n exponentially small under normal conditions) probability of a sudden discrete transition.
Make sense?
Make sense?
Yes, this part was obvious! What I meant with those bit-flips was the exponentially small probability of a sudden discrete transition. Why could accidental transitions like this not accumulate? Because they are selected against fast enough? I wouldn't expect those epigenetic marks to have enough redundancy to reliably last to the end of an organisms' lifetime, because methylated cytosine is prone to deaminate (which is why CG is the least frequent 2-mer at a ~1% frequency, rather than the ~6.25% you'd expect on baseline). I am confused how the equilibrium works here, but it seems like mutational load could explain why organisms who rely on methylating cytosine have less CG's than would be useful to maintain epigenetic information. Things would be different in organisms that don't rely so heavily on methylating cytosine.
So you're saying that the persistent epigenetic modification is a change in the "equilibrium state" of a potentially methylated location?
Does this mean that the binding affinity of the location is the property that changes? i.e. all else being equal, a location with high affinity will be methylated much more often than a location with low affinity, because the methyl groups will tend to stick harder or longer in the high affinity location.
But if that's the case, it seems like there still must be some persistent structural feature responsible for setting the binding affinity to high or low...
So you're saying that the persistent epigenetic modification is a change in the "equilibrium state" of a potentially methylated location?
Yes.
Does this mean that the binding affinity of the location is the property that changes?
Not quite, it would most likely be a change in concentrations of enzymes or cofactors or whatever which methylate/demethylate the specific site (or some set of specific sites), rather than a change in the site itself.
Ask an epigenetics researcher what they study, and the standard story you'll hear goes something like this...
"Sometimes a little methyl group (i.e. -CH3) gets stuck on the side of a strand of DNA. Turns out these guys are pretty important! They're copied over when cells replicate, so they stick around long-term, and they can activate or repress (usually repress) nearby genes on the DNA strand. In particular, different types of cells all have the same DNA code, but something has to be different in order for the cells to "remember" what type they are and behave differently. And sure enough, those methyl modifications differ across cell types! They're like an extra information storage mechanism, on top of the DNA, which can encode things like cell type and make different cell types behave differently, among other forms of memory."
That story is wrong. Many of the details are correct, but there's one crucial mistake, and once we correct that mistake we end up with a very different mental picture.
The mistake: methyl groups usually do not stick around long-term; they turn over regularly. Here are two studies which measured the turnover. Turnover timescale varies by location on the DNA strand, but turnover every few days is typical.
With that in mind, here's how I'd describe the way methyl modifications actually work...
"Sometimes chemical signals have multiple steady states - for instance, maybe A suppresses B and B suppresses A, such that both (high A, low B) and (low A, high B) are stable. Turns out chemical subsystems with multiple steady states are pretty important! Since the state is steady, it can stick around long-term, and even stick around when a cell replicates. In particular, different types of cells all have the same DNA code, but something has to be different in order for the cells to "remember" what type they are and behave differently. Subsystems with multiple steady states play that role! They're like an extra information storage mechanism, on top of the DNA, which can encode things like cell type and make different cell types behave differently.
And it turns out that methyl modifications, along with things like proteins and small molecules and all the other typical chemical types in a cell, are among the chemicals which can be part of such a subsystem. In fact, methyl modifications are particularly well suited to this role, because they can be highly redundant at relatively low metabolic cost: there can be methyl modifications at many sites which all 'say the same thing' about the cell state, making the memory quite robust!"
Personally, I ran into this while studying aging. If we imagine that methyl groups stick around indefinitely, then (at least in long-lived cells) they're a prime candidate for mediating age-related changes. But if the methyl groups are instead part of a dynamic equilibrium, especially with high redundancy (and therefore stability), then that's a whole different situation.